The pipeline in detail
The following detailed description is only meant for advanced users.
Psitblastn
Selenoprofiles uses psitblastn from the NCBI blastall package. This program can be considered an extension of tblastn. Instead of using only a single sequence as query, it considers also a Position Specific Scoring Matrix (PSSM). This allows to utilize the relative proportions of allowed residues at each profile position. Normally, its more famous relative psiblast (extension of blastp) is used iteratively against a sequence database, building a PSSM with the matches it finds. In our use of psitblastn, no iteration at all is performed, since the profile alignment is already provided as input and the PSSM can readily be derived.
Pre-clustering
We experienced that when a profile is very broad (i.e., contains sequences quite dissimilar to each other), the psitblastn search is not very sensitive. For this reason, selenoprofiles implements a procedure that analyzes the input profile alignment in terms of its variability, and clusters its sequences based on their sequence identity. If the profile has a high variability, then this procedure will produce more than one cluster.
Then, a psitblastn search for each cluster is performed: one PSSM is built from the sequences of each cluster. Consequently, often there are overlapping blast hits coming from the searches of different clusters. Those are merged, keeping only the best one for each overlapping set. The sequence identity threshold for the profile clustering procedure can be defined for each profile (clustering_seqid parameter), or goes to the default value defined in the main configuration file.
Consensus blast query
Psitblastn build a PSSM along the positions of a certain sequence of the profile, elected as the blast query. In our experience, the choice of the blast query has a big effect on the results of the search. The blast query is built for each search, as a “consensus”. Its sequence is given by the most present amino acid at each position of the alignment (or of the cluster, if more than one is present). There are two exceptions to this. For selenoproteins, in the positions where at least a Sec is detected, the blast query always bears a U. Then, the positions featuring a lot of gaps in the alignment are skipped. The maximum percentage of gaps for a column depends on the option max_column_gaps_for_blast_query, either specified in the profile configuration or set to the default in the main configuration file.
For technical reasons, all blast hits loaded in selenoprofiles are transformed so that their alignments are between the target and a unique query sequence, named the master blast query. This allows to have a more homogenous kind of data for subsequent computation: otherwise, blast hits coming from different clusters searches would have different sequences as query.
Merging exons by co-linearity
After the overlapping hits from the various cluster searches are removed, blast hits are once again analyzed, and those likely to be exons of the same gene are joined: they are merged by co-linearity. This means that if a blast hit is downstream of another one, and also the correspondent portions of the aligned query sequences are one downstream of the other in the same direction, the blast hits will be merged into a single object (if they are not too far away). This procedure is done to minimize redundant computation.
Blast filtering
Blast hits are filtered according to criteria that may be specified for each profile. In our experience, different protein families need very distinct criteria. Some families typically match a lot of spurious hits, while some others need loose filters to find all results. All filtering procedures in selenoprofiles are written in python and can be customized by the user, utilizing a set of methods that are already provided or can be created by the user. Filtering is detailed in a later. Blast filtering is performed actually before removing redundancy across cluster searches, and also before merging by co-linearity. This is because merging blast hits requires loading them all into memory, sorting them and parsing them – which sometimes would take very long if all blast hits in a output file are considered.
If for some reason you want to inspect manually the blast hits passing the filter, you can use option -filtered_blast_file and provide a file as argument, which will be created. Blast hits within this file have not been subject to inter-cluster or co-linearity merging.
Maximum number of blast hits
In selenoprofiles, the computation is largely dependent on the number of blast hits passing filtering. For this reason, there is a fixed maximum number of blast hits which can be considered. The default value is extremely loose: 2500. When the limit is passed for a family, a warning is printed on screen and the workflow follows keeping only the blast hits found so far. Blast hits are read in the order they are in the blast output file. Blast sorts the hits according to the chromosomes (or contigs) they are located on, ordering the chromosomes according to the e-value of the best HSP found on them. This way of sorting is not strictly best-to-worse but it is similar, therefore most likely you won’t lose any bona-fide gene because you reached the maximum limit of blast hits.
Also, the blast outputs produced searching the different clusters are read in order, with the cluster containing the highest number of sequences being first. Therefore, the first blast output read should be the most representative.
In an older version of selenoprofiles, the computation would simply stop if the max number of blast hits is reached. This behavior can be restored by setting off the relevant option, with -blast_filtering_warning 0.
Exonerate
Each alignment coming from the blast phase is used as a seed to run exonerate in the corresponding genomic region, using a proper extension procedure.
Reading and joining exonerate predictions
Exonerate is run on a chromosomic region in which a blast hit was found, and typically it will give a single gene structure prediction in output. Nonetheless, this is not always the case. For this reason, selenoprofiles considers only the exonerate prediction which, among those in its output file, overlaps with the blast hit used as seed. If more than one overlapping prediction is present (very rarely), the best scoring one is taken.
Also, exonerate generally joins the exons belonging the same gene, including the prediction of splice sites. Nonetheless, often no good scoring splice sites are found and such predictions may be found separated. Selenoprofiles attempts to merge the “main” exonerate prediction with the others in the same file, using the co-linearity concept previously mentioned for blast hits. This behavior is turned off when selenoprofiles option no_splice is active.
Cyclic exonerate
Exonerate is run through a peculiar routine called cyclic exonerate (see figure below; see also selenoprofiles paper). This procedure comes in response to the following problem: if we want to run exonerate on a certain genomic region where a blast alignment gave us the hint of an homology match, we need to decide the boundaries of the region searched by exonerate. Of course the region provided by blast needs to be extended, but by how much? Gene sizes are incredibly variable. Taking the biggest size ever observed would result in a huge amount of useless computation, while on the other side taking an average would obviously be inappropriate for a fraction of cases.
This routine solves this problem by running exonerate more than once, increasing progressively the genomic space searched on both sides by a fixed parameter. The cycle stops when a run predicts the same coding sequence of the previous one. If the extension parameter is chosen greater than the largest expect intron, the procedure ensures that the widest prediction possible is achieved.
The cyclic routine runs exonerate on average less than 3 times. Given the high speed of exonerate, this is more than acceptable timewise, also considering that this step is not the most computationally intensive in selenoprofiles. Also, if the chromosome (contig) is comparable in size to the extension parameter, the cyclic routine is not performed and the whole chromosome is used as target. The default exonerate_extension is 200.000 bases.
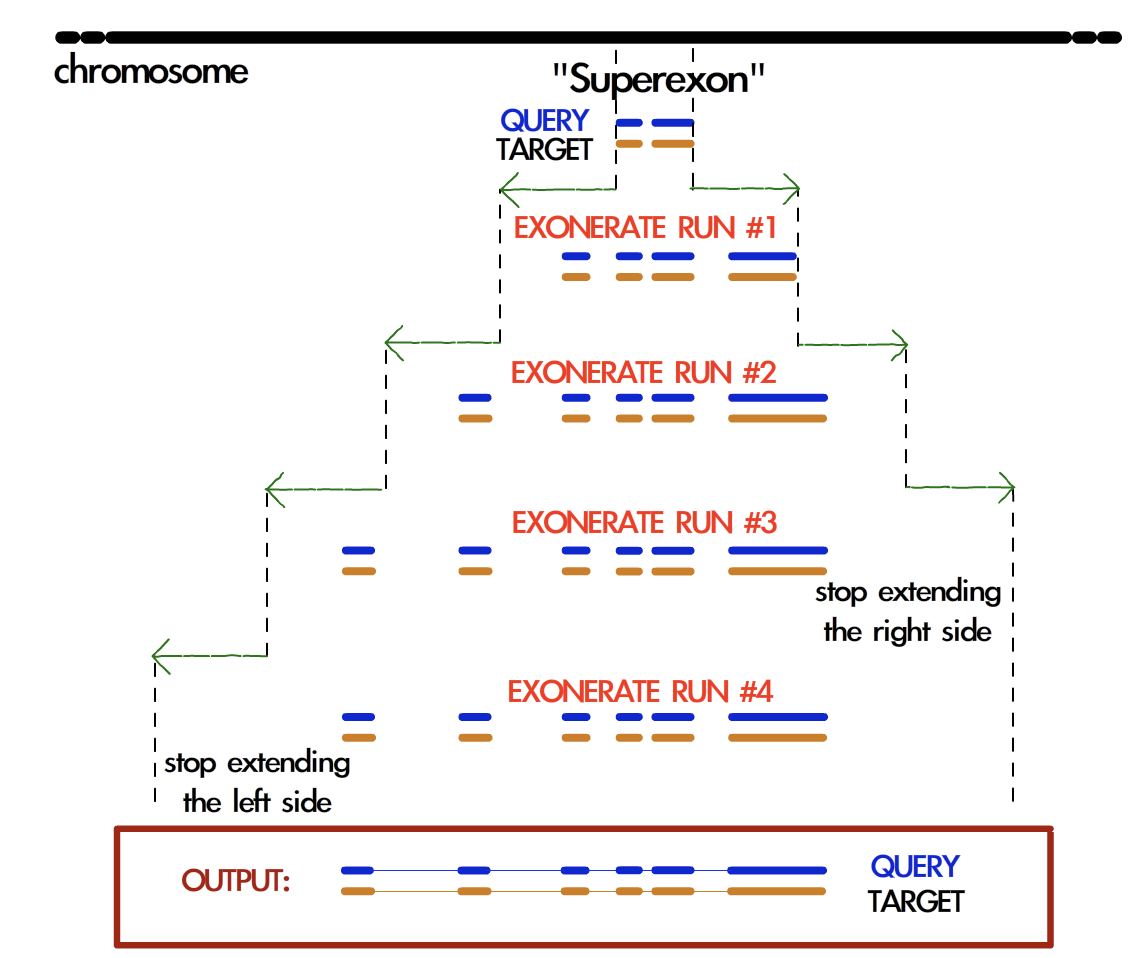
Schema of the cyclic exonerate routine, from the selenoprofiles 2010 paper. A “superexon” represents either a blast hit, or more than one merged by co-linearity.
Choosing the best query from the profile
Exonerate accepts a single sequence query, but in the pipeline the information of a whole profile of sequences is available. Thus, selenoprofiles chooses the best query sequence in the profile for each candidate gene, by searching the query which is most similar to the sequence predicted in the target. To do so, the current predicted sequence is mapped to the profile alignment exploiting the query, which is in common between the prediction alignment and the profile alignment. This is done at every cycle, before running exonerate. At the first run, the predicted sequence in the target is given by the blast prediction, and for each subsequent run is given by the previous exonerate prediction. Before closing the cyclic routine, it is checked that the best query is still the one that was lastly chosen, otherwise one more cycle is run.
Modifying exonerate behavior for selenocysteine sites
Selenoprofiles was created to predict genes belonging to selenoprotein families. It is able to do so by using special scoring schemes with exonerate and genewise (blast is used with a neutral score at these sites).
When dealing with Sec families, a particular scoring matrix derived from BLOSUM62 is used: the alignment of a “*” character to a stop codon in the target is scored positively. When the query is chosen from the alignment, its sequence is modified before it is used by exonerate: all the positions which contains at least one Sec in the profile are changed to “*”, favoring de facto the alignment of Sec positions to UGA codons.
Removing redundant exonerate hits
Often, blast hits representing exons of the same genes pass through the co-linearity merge procedure previously described, mainly because this is kept with loose parameters to avoid joining accidentally similar, close genes. When this happens, such blast hits are used to seed cyclic exonerate runs which end up in identical gene structure predictions.
After all exonerate runs are computed, their predictions are analyzed and the redundant ones are dropped, to save computational time in the genewise phase.
Genewise
Generally, genewise represents the most computationally expensive step in selenoprofiles, together with blast. Genewise performs a tblastn-like alignment complemented with prediction of splice sites, basically just like exonerate, which. Nonetheless, genewise does not use heuristics and its running time is considerably higher. When you need to maximize speed, you can skip the genewise step using option -dont_genewise.
Genewise is generally run on genomic regions defined by an exonerate prediction, attempting to refine them. Such genomic regions are extended by a parameter, genewise_extension, which is only 100 bases by default. Unlike exonerate, genewise is run only once.
Genewise “to be sure” routine
In many cases exonerate does not produce any prediction in output. This happens particularly for very low scoring blast hits, which cannot be reproduced by exonerate. In these cases, selenoprofiles performs a genewise routine called “to be sure”, in which a blast hit (instead of an exonerate prediction) is used as seed of a genewise run. In our experience this rescues many predictions, but it is very computationally expensive. The extension of genomic region in the blast hit is defined by the genewise_tbs_extension parameter, which is 10.000 bases by default. One can avoid running this routine using option -genewise_to_be_sure 0.
The query in genewise
As for exonerate, a single query sequence needs to be chosen to be run with genewise. In a standard run, the same query used by exonerate is chosen, as this is already the most similar to the target sequence. When a blast hit is used in the genewise “to be sure” routine, the best sequence is chosen from the profile by maximizing identity with the target, in the same way it is done in the first cycle of an exonerate routine.
Modifying genewise behavior for selenocysteine sites
For genewise, a trick similar to the one described for exonerate is used when searching for selenoprotein families. Each query used is modified to bear a selenocysteine (“U”) corresponding to every column of the alignment which possesses at least one. Then, the translation table normally used by genewise is changed, using one in which UGA is translated as “U”. The scoring matrix given to genewise is then a modified BLOSUM62, in which a “U” in the target is score positively only to a “U” in the query.
Improving predictions
In selenoprofiles a few steps are dedicated to the processing of the predicted gene structures, in order to correct them. All of them are implemented as methods of the superclass p2ghit, which comprises the classes for blast, exonerate or genewise predictions (see p2ghit class in Advanced usage). These methods are run through “Actions” (also explained in Advanced usage) specified in the main configuration file. You can turn off the improvements methods by removing, or commenting (with #), the corresponding lines in the main configuration file.
The first improvement is called remove_internal_introns and is performed only on blast hits. This method is useful because often blast joins two or more coding exons in a single hit, when the exons are on the same frame and the resulting stretch of unaligned amino acids in the target is acceptable in terms of scoring. A typical blast hit containing an evident intron is shown here:
Score = 100 bits (249), Expect = 4e-20
Identities = 49/93 (52%), Positives = 59/93 (63%), Gaps = 26/93 (27%)
Frame = +2
Query: 12 LEPYMDENFITRAFAKMGENPVSVKLIRNKMTG--------------------------E 45
LEPYMDENFI+RAFA MGE +SVK+IRN++TG
Sbjct: 103916 LEPYMDENFISRAFATMGELVLSVKIIRNRLTGYV*SLFVFYHIPNFGVHLHTLFSLSRI 104095
Query: 46 PAGYCFVEFADEASAERAMHKLNGKPIPGANPP 78
PAGYCFVEFAD A+AE+ +HK+NGKP+PGA P
Sbjct: 104096 PAGYCFVEFADLATAEKCLHKINGKPLPGATPV 104194
The portion YV*SLFVFYHIPNFGVHLHTLFSLSRI is the translation of an intron.
It has no correspondence in the query, and it also contains a stop codon
(it is normal as introns have no coding constraint). The
remove_internal_introns method detects these cases by searching the
sequence in the target for stretches of at least 18 bp (6 amino acids)
not aligned to the query, and removes them from the prediction.
The second improvement is performed by function clean_inframe_stop_codons. This is applied to predictions by all programs, and comes from the observation that often these programs include stop codons that should be avoided. This would cause these predictions to be mislabelled as pseudogenes. This method is simple in principle: it checks for the presence of stop codons close to exon boundaries (default maximum: 10 codons). If it finds any, it removes the stop codons and also the portion which links it to the closest exon boundary.
The third improvement is exclude_large_introns. This is particularly useful on exonerate predictions, which sometimes possess extremely large introns, due only to spurious similarity with far away regions, and to the presence of decent splice sites just by random. This function detects each such large intron (default >= 140000 nt), and removes all exons (typically just one) at one side of that intron, the side with the smallest coding sequence.
While all described methods are applied before prediction choice, the fourth and fifth improvements are performed at the end of pipeline, only on the predictions passing the filter; nonetheless, they are described here below.
The functions complete_at_five_prime and complete_at_three_prime are attempts to complete the coding sequence predictions looking for an upstream ATG and a downstream stop codons. Let’s see the corresponding lines in the selenoprofiles.config file:
ACTION.post_filtering._improve4= if x.filtered=='kept': x.complete_at_three_prime(max_extension=10, max_query_unaligned=30)
ACTION.post_filtering._improve5= if x.filtered=='kept': x.complete_at_five_prime(max_extension=15, max_query_unaligned=30, full=False)
The completion at 5’ is performed only if a ATG is found before a stop codon, and if at most 15 codons would be added. Also, two other conditions must be met: no non-standard characters must be find in the 5’ extension, and the profile query of this prediction must have an unaligned portion at N-terminal not bigger than 30 amino acids. This is to avoid completing partial hits, whose upstream ATG are not likely to be the real starts, as other large portion of coding sequence are expected upstream.
Also, normally the function stops when the first methionine is found upstream – if the first codon is already a AUG, no extension is performed. When full=True is provided, it attempts instead to extend to the furthest possible methionine, when coupled with high values of max_extension.
The completion at the 3’ is performed only if the profile query has an unaligned portion at C-terminal not bigger than 30 amino acids, if the extension is at most 10 codons, and if no strange characters are found in the candidate extension.
The use of these two methods is very limited by default, because selenoprofiles is meant to kept its nature of homology-based tool. However, their extent can easily be altered by the user through the main configuration file, as shown earlier in “searching RNA sequences or bacterial genomes”.
Selenoprofiles can be customized to perform additional improvements. The user has to write a function accepting a p2ghit as input, and modify the main configuration file to run the function at the right step, using actions.
Prediction program choice
After the genewise step, three predictions are available for every candidate: one by blast, one by exonerate, and one by genewise. The predictions are analyzed and only one is taken to represent this candidate gene to the filtering phase, and possibly to output. The function choose_prediction is used to decide among any number of candidates. This same function is used during all steps in which genes are merged to remove redundancy, to decide which one to keep. The following conditions are checked in order: if at any point only one of the predictions shows to be better than all others for a criteria, the function stops and that prediction is returned.
The first condition checked is the presence of frameshifts. If a prediction possesses frameshifts while another doesn’t, the latter is taken.
Then, if the predictions come from a selenoprotein family, the number of aligned Sec positions is considered: if one possess more than the others, it is chosen.
The number of in-frame stop codons (others than SecTGAs) is then checked: if one possess less than the others (for example one has none, while the others have), it is chosen.
After, the length of the predicted coding sequence is determinant: the prediction featuring the longest sequence is chosen.
If at this point the choice has not been made yet, the prediction whose program has highest priority is chosen, given these priorities in descending order: genewise, exonerate, blast.
Option -no_blast forces selenoprofiles to choose the exonerate or genewise prediction. This is useful only if an accurate splice sites prediction is important for you. It comes at the cost that, when only the blast prediction is available (for example because exonerate produced an empty output, and genewise an invalid alignment), the candidate is always discarded.
Labeling
After a single prediction per candidate is chosen, this is analyzed and labelled.
For standard families, there are only two possible labels: homologue (a regular prediction) and pseudo (with any in-frame stop codon or frameshift). It is possible for the user to define its own labeling procedure: this is shortly described later (“option -add”).
For selenoprotein families, labeling is used to characterize the amino acid aligned to the Sec position. Generally there’s a single Sec in selenoproteins. If there’s more than one, the label assigned by selenoprofiles depends on the most-left aligned Sec position. The possible labels are selenocysteine, cysteine or any other amino acid (only rarely found at these positions though). If the prediction does not span any Sec position, it is labelled as unaligned. If it contains frameshifts or in-frame stop codons (apart from Sec-TGA), then it is labeled as pseudo. An additional label, uga_containing, is assigned to those predictions whose only pseudogene feature is one or more in frame UGAs (of course not aligned to Sec positions). This label is useful because very rarely the scoring schemes used for selenoprotein families allow the alignment over a non-Sec UGA, and we don’t want to filter those out as if it were pseudos. Also, the label may be useful to discover new Sec positions in known selenoprotein families.
Final filtering
After labeling, predictions are evaluated through the final filter before output. This filter, exactly as the blast filter, can be specific for each family and be written using the methods provided in selenoprofiles classes. The filter outcome is summed up in a filtering label, hereafter called “filtering state” (or just state) to differentiate it from the label assigned in the previous step. The final filter actually consists of two separate filters, called p2g_filtering and p2g_refiltering in the configuration files. A prediction excluded by the first one will be assigned a state of filtered. A prediction excluded by the second one will be assigned a state of refiltered.
Just before the predictions enter the final filter, there is an additional redundancy check: the predictions overlapping each other are compared and only the best one is kept. Predictions discarded this way are assigned a state of redundant.
Those predictions which passed all the redundancy check and the two steps of the final filter without being discarded are assigned a state of kept and represent the normal output of selenoprofiles.
Nonetheless, the user may decide to output the predictions with a different state, using the -state option, optionally with multiple arguments, comma separated with no space within. If for example you want to output all filtered and refiltered predicted, add to your command line:
-state filtered,refiltered
The -state option can accept the following arguments: kept, filtered, refiltered, redundant or overlapping (see below). There is a way to have even more control on what prediction are output: the -output_filter option. This accepts a procedure with the same syntax of filters and actions, which is evaluated for every prediction: those for which this evaluates to True will be output. If for example you want to output only predictions on the positive strand, you can use:
-output_filter "x.strand==‘+'"
To do this, you need to know a bit about the classes used in selenoprofiles, described in the Advanced usage section. After filtering, results are stored in the sqlite database, ready for the output phase (below).
Removing inter-family redundancy
Selenoprofiles scans for multiple profiles in a single run. The output is produced only when all families have been searched. This is because results from different profiles may overlap, especially when some of them share a certain degree of sequence similarity. So after all results are stored in the database, this is parsed and every prediction is compared with all others on the same chromosome (or contig). When two such predictions overlap, the function choose_among_overlapping_p2gs_interfamily is used to decide which one to keep. The other is assigned a state of overlapping. These predictions will not be output by default. Note that this operation is performed directly on the database: the intermediate text files written in the filtering phase will display the state previously assigned.
Another important note: the inter-family redundancy check is performed every time an output phase is run, and depends on the results present in the database at that moment. For this reason, searching several profiles in distinct selenoprofiles runs will lead to more (or the same number of) output files than searching all of them in a single run. The results database at the end will be identical, but as when every profile reached its output phase, the predictions of all other profiles were not available, the inter-family redundancy cannot be checked properly.
If you searched different profiles on separates runs, the best thing to do is just delete all output files and rerun selenoprofiles with all these profiles using -D flag to re-run database storage. No heavy computation will be repeated, and only the output files for the non-overlapping predictions will be produced.